We couldn’t survive without proteins. They’re essential molecules that provide cells with structure, aid in chemical reactions, support communication, and much more. Portion out protein numbers with us below!
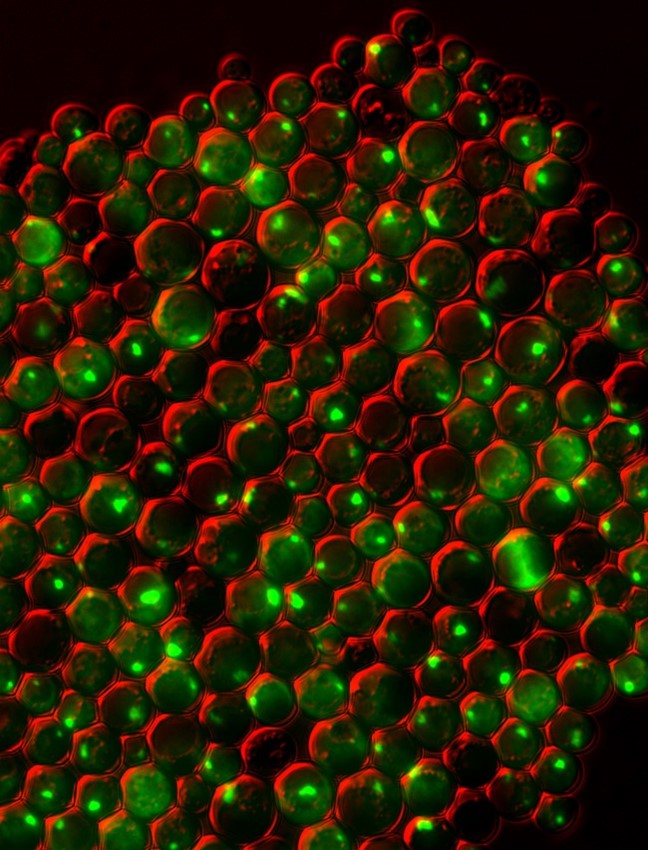
These green spots are clumps of protein inside yeast cells that are deficient in both zinc and a protein that prevents clumping. Credit: Colin MacDiarmid and David Eide, University of Wisconsin at Madison and the Journal of Biological Chemistry.
10 Trillion
That’s how many proteins scientists estimate are in each human cell.
229,378
That’s how many structures researchers shared with the scientific community through the Protein Data Bank (PDB) from its establishment in 1971 to the end of 2024. The PDB is a global repository for 3D structural data of proteins, DNA, RNA, and even complexes these biological molecules form with medicines or other small molecules.
42
That’s the percent of your body weight (not counting water) that’s made up of proteins.
“I found a passion for both biology and chemistry in high school and thought, Well, that must mean I’m a biochemist! Luckily my naïve thought was correct. I am a biochemist,” says Bil Clemons, Ph.D. He’s a professor of biochemistry at the California Institute of Technology (Caltech) in Pasadena, where he’s been teaching and running a lab for nearly 20 years.
A Path to Research
Dr. Clemons doesn’t remember a time when he wasn’t interested in science or curious about the world. “I think, fundamentally, that’s what being a scientist is: being curious about how the world works,” he says. As a child, he’d open seed pods to see the insides or take toys apart to see how their tiny motors worked. He couldn’t always figure out how to put the toys back together, though, which led to his parents warning him not to ruin his siblings’ new toys on Christmas morning.
Anesthesia is a treatment that prevents patients from feeling pain during procedures like surgery, medical tests, and dental work. Anesthesiologists are doctors who have been specifically trained to give medicines used for anesthesia, which are called anesthetics.
Depending on the procedure they’re having, patients receive different types of anesthesia:
Haley Bridgewater, a graduate student at Boise State University in Idaho, is sure she wants to continue studying infectious diseases after she graduates with her Ph.D., but she’s finding it difficult to choose a specific topic within that branch of biomedical science. “My problem is that I like them all. The more I look into specific research topics to narrow down my options, the longer my list of potential topics grows,” she says.
Haley Bridgewater in front of the Boise River on Boise State University campus. Credit: Elise Overgaard, Ph.D., Boise State University.
Haley’s early introduction to science wasn’t related to the biological sciences at all. She grew up in Los Alamos, New Mexico, where her dad studied nuclearchemistry. Discussions about chemistry, physics, and rockets surrounded her, and she would often stare up at the night sky to catch a glimpse of a meteor shower or the International Space Station passing by. But she was even more curious about what was below her feet: What makes an insect different from a rock? What does the microscopic world look like? She received a microscope one year for her birthday and carried it with her everywhere so she could try to answer these questions.
Global Experiences
Haley took an advanced biology class in high school, where she learned not only about the living world, but also the many exciting scientific careers available, such as becoming a researcher. She moved to Tacoma, Washington, and earned a bachelor’s degree from Pacific Lutheran University (PLU), where she majored in biology and global religion.
“Curiosity was a central theme in my learning process,” says Sudha Chakrapani, Ph.D., a professor and chair of the department of pharmacology at Case Western Reserve University in Cleveland, Ohio. As a high schooler in India, she especially enjoyed her science classes because they fostered her curiosity and allowed her to ask more questions than other subjects did. She was curious about how to use science to solve the challenges she and her community faced, like access to safe drinking water. Seawater surrounded them, so could they find a way to convert it into drinking water?
As part of India’s annual National Teachers’ Day celebration, high school seniors take on the role of educators and teach their younger peers for the day. Dr. Chakrapani loved the experience, and it solidified what she already knew: She wanted to go to college to be a science teacher. After earning her bachelor’s degree, she entered back-to-back master’s programs in biochemistry and biomedical engineering, where she had the opportunity to do hands-on research.
Numbers are everywhere in chemistry. You can’t balance equations, determine limiting reactants, or calculate percent yields without them. So, let’s dive into some of the significant figures in chemistry!
3
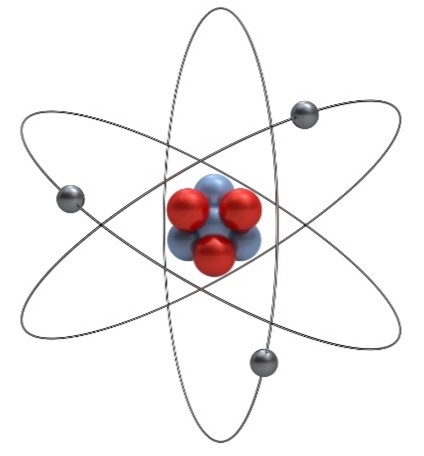
A lithium atom with three protons (red) and four neutrons (blue) in the nucleus and three gray electrons orbiting around them. Credit: iStock.
That’s the number of different types of particles—protons, neutrons, and electrons—that make up atoms, the basic unit of all matter. Protons are positively charged, neutrons are neutral, and electrons are negatively charged. The number of protons in an atom determines what element it is, and atoms usually have an equal number of protons and electrons. Atoms can have different numbers of neutrons, though, and atoms with the same number of protons and different numbers of neutrons are called isotopes. Protons and neutrons make up the core—or nucleus—of an atom, and electrons orbit around them.
4.9 Million
That’s how many miles per hour the electron in one hydrogen atom in a molecule of water is moving. At that rate, the electron could make it from New York City to Los Angeles in about 2 seconds!
It might sound like a science fiction author made up genetic engineering, but it’s a real tool researchers use in the laboratory! A gene is a segment of DNA that codes for a protein. The information within a gene directs the building of a protein, block by block, through the process of gene expression. For a variety of reasons, including learning about certain cellular processes, scientists use genetic engineering in the lab to manipulate a cell’s genes and the proteins they encode.
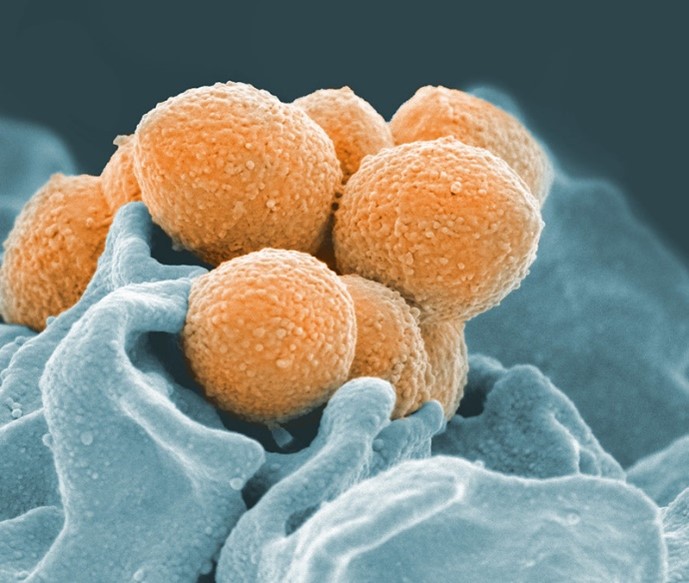
Streptococcus bacteria under the microscope. Species in this genus, such as Streptococcus pyogenes, have a CRISPR-based defense system. Credit: NIAID/NIH.
Eventually, researchers discovered that these sequences are part of a bacterial immune system. (Just like humans, bacteria are susceptible to viral infections!) Some bacteria are able to insert short sequences of DNA from viruses that previously infected them into their own genome, allowing them to “remember” and more quickly recognize that virus in the future. If the invader tries to attack again, the bacterium recognizes and kills it by chopping up the part of its DNA that matches the “memory” using a special type of protein, an enzyme called CRISPR-Associated (Cas) protein. Our own immune systems also have the ability to remember pathogens through our adaptive immune response.
“Science has always impacted me, but I didn’t realize how much until I actually became a scientist,” says Elias Picazo, Ph.D., assistant professor of chemistry at the University of Southern California in Los Angeles. We talked to Dr. Picazo about his path to becoming a scientist, some of the challenges he faced along the way, and his research inventing new ways to make chemical bonds.
Get to Know Dr. Picazo
Books or movies? Movies
Beach or mountains? Mountains
Favorite music genre? Pop
Rainy or sunny? Sunny
Salty or sweet? Sweet
Music or podcast? Podcast
Washing glassware in the lab or dishes in your kitchen? Glassware
Have you wondered what controls the most basic functions of our bodies, like breathing, moving, and sleeping? Chemicals called neurotransmitters play a central role. Neurotransmitters pass messages from one nerve cell to another, and sometimes to muscles or glands. These messages may:
Prompt the next nerve cell to pass on the message, prevent the message from going any further, or adjust how the message is passed on
Cause a muscle to contract, like our intestines do when they digest food
Colton Pelletier with Roti-Bot. Credit: Grace Boland, RWU.
During his time at Roger Williams University (RWU) in Bristol, Rhode Island, Colton Pelletier built a robot that will help simplify data collection for research projects in the lab he worked in—and others—for years to come. Aiding in Colton’s success in the lab was NIGMS funding through the Institutional Development Award (IDeA) Networks of Biomedical Research Excellence (INBRE) program. INBRE funds statewide networks of higher education in IDeA states such as Rhode Island, which have historically received low levels of NIH funding. The program supports faculty research, mentoring, student participation in research, and research infrastructure by connecting primarily undergraduate institutions with research-intensive universities in the state.