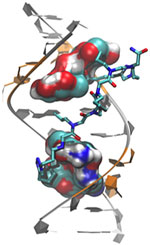
Myotonic dystrophy type 2 (DM2) is a relatively rare, inherited form of adult-onset muscular dystrophy that has no cure. It’s caused by a genetic defect in which a short series of nucleotides—the chemical units that spell out our genetic code—is repeated more times than normal. When the defective gene is transcribed, the resulting RNA repeat forms a hairpin-like structure that binds to and disables a protein called MBNL1.
Now, research led by Matthew Disney of The Scripps Research Institute (TSRI), Florida Campus, has revealed the detailed, three-dimensional structure of the RNA defect in DM2 and used this information to design small molecules that bind to the aberrant RNA. These designer molecules, even in small amounts, significantly improved disease-associated defects in a cellular model of DM2, and thus hold potential for reversing the disorder.
Drugs that target toxic RNA molecules associated with diseases such as DM2 are few and far between, as developing such compounds is technically challenging. The “bottom-up” approach that the scientists used to design potent new drug candidates, by first studying in detail how the RNA structure interacts with small molecules, is unconventional, noted Jessica Childs-Disney of TSRI, who was lead author of the paper with Ilyas Yildirim of Northwestern University. But it may serve as an effective strategy for pioneering the use of small molecules to manipulate disease-causing RNAs—a central focus of the Disney lab.
This work also was funded by NIH’s National Cancer Institute.
Learn more:
The Scripps Research Institute News Release ![]()
Disney Lab