
Proteins (such as hemoglobin, actin, and amylase) are workhorse molecules that contribute to virtually every activity in the body. Some of proteins’ many jobs include carrying oxygen from your lungs to the rest of your body (hemoglobin), allowing your muscles to move (actin and myosin), and digesting your food (amylase, pepsin, and lactase). All proteins are made up of chains of amino acids that fold into specific 3D structures, and each protein’s structure allows it to perform its distinct job. Proteins that are misfolded or misshapen can cause diseases such as Parkinson’s or cataracts.
While it’s straightforward to use the genetic code to predict amino acid sequences of proteins from gene sequences, the vast diversity of protein shapes and many factors that influence a protein’s 3D structure make it much more complicated to create simple folding rules that could be used to predict proteins’ structures from these sequences. Scientists have worked on this problem for nearly 50 years, and NIGMS has supported many of their efforts, including the Critical Assessment of Structure Prediction (CASP) program.
Accurately predicting protein structures could benefit science and human health in several ways. It could help researchers to:
- Develop medicines by allowing them to predict what diverse shapes of proteins in the body look like and how potential drugs will interact with them
- Design proteins for specific tasks, such as detecting toxins or neutralizing viruses
- Learn how the shapes of proteins that are altered by genetic mutations differ from shapes of normally occurring proteins in the human body, which might enable development of medicines to correct the effect of these mutations
Although experiments can be used to determine protein structures, they are much more time consuming and expensive than computational methods. So, in the future, researchers can hopefully use computers to accurately predict protein structure and use the predicted structure to, for example, model how a large number of potential drugs will interact with the protein. The time-consuming experimental methods would be used to evaluate only the most promising candidates predicted by the computation.
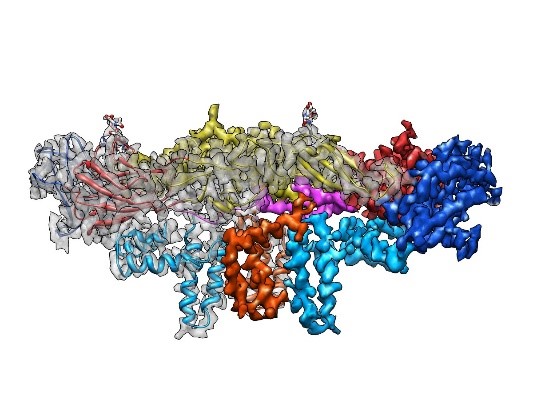
In addition to the time and cost involved with more traditional lab experiments, despite much effort, such experiments sometimes fail to determine a protein’s structure. Accurate computer predictions could provide the clues needed to decipher some of these challenging structures.
Every 2 years since 1994, CASP has held a competition where teams of researchers test computational methods they’ve developed to predict protein structures from their amino acid sequences. The most recent competitions have involved about 100 teams worldwide. To assess the methods’ accuracy, impartial evaluators compare the structures the teams predict with structures determined using lab experiments. The average accuracy of the prediction methods has increased with every CASP competition.
“The impact of CASP is essentially bringing rigor into the field,” says Krzysztof Fidelis, Ph.D., the director of the Protein Structure Prediction Center at the University of California, Davis, and a founder of CASP. The competition tests methods objectively and encourages continual improvements.
An Important Milestone and Future Goals
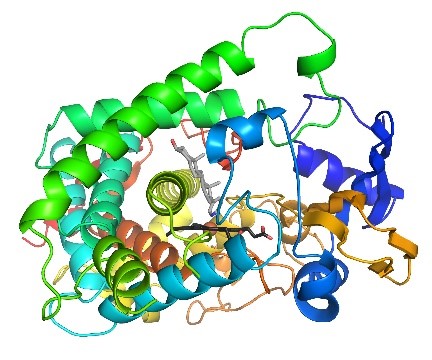
In 2020, a method tested in CASP appeared to be highly accurate for predicting the structures of many proteins or domains—distinct structural or functional units within proteins—that are made from a single amino acid chain. This method, called AlphaFold, is noteworthy for its use of artificial intelligence (AI), a technology with a growing role in protein structure prediction. The researchers who developed AlphaFold were inspired to build an “intuitive” AI in part by seeing people play Foldit, a computer game that allows the public to contribute directly to scientific research by puzzling out potential shapes for proteins.
The CASP organizers say that the AlphaFold advance builds on achievements made by many teams in previous CASP rounds. A next goal for CASP competitors is predicting the structures of protein complexes, which are made up of multiple amino acid chains and include most human proteins. The researchers also hope to eventually model proteins’ conformational states and dynamics—the ways the proteins change shape in response to environmental factors and time.
Inspiring Other Efforts
One of the most important features of CASP is that it brings together a community of researchers to pursue a common goal and build upon one another’s work. The competition has inspired similarly structured efforts, such as the Critical Assessment of PRediction of Interactions (CAPRI) and Critical Assessment of protein Function Annotation algorithms (CAFA) programs.
CAPRI is a community-wide experiment that enables researchers to test methods for predicting how proteins interact with one another or with other molecules, and CAFA is a competition where scientists test methods for predicting a protein’s function based on its amino acid sequence. Efforts like these and CASP help researchers rigorously and objectively evaluate computational methods and propel protein science forward in ways that could ultimately improve human health.
CASP is supported by NIGMS grant R01GM100482.