Did you know that kids aren’t the only ones playing around in sandboxes? The term sandbox may evoke a childhood memory of sensory play, but it’s also used to describe a virtual environment where someone can learn from digital products.

Biomedical Beat Blog – National Institute of General Medical Sciences
Follow the process of discovery

Did you know that kids aren’t the only ones playing around in sandboxes? The term sandbox may evoke a childhood memory of sensory play, but it’s also used to describe a virtual environment where someone can learn from digital products.


The power of computer code has been a longtime fascination for Tomas Helikar, Ph.D., a professor of biochemistry at the University of Nebraska-Lincoln (UNL). In college, when he learned he could use that power to help researchers better understand biology and improve human health, Dr. Helikar knew he’d found his ideal career. Since then, he’s built a successful team of scientists studying the ways we can use mathematical models in biomedical research, such as creating a digital replica of the immune system that could predict how a patient will react to infectious microorganisms and other pathogenic insults.
A Career in Computational Biology
Dr. Helikar first became involved in computer science by learning how to build a website as a high school student. He was amazed to learn that simple lines of computer code could be converted into a functional website, and he felt empowered knowing that he had created a real product from his computer.
Continue reading “Building a Digital Immune System”

Adam Gormley, Ph.D., describes himself as a creative and adventurous person—albeit, not creative in the traditional sense. “Science allows me to be creative; to me, it’s a form of art. I love being outdoors, going on sailing trips, and spending time adventuring with my family. Research is the same—it’s an adventure. My creative and adventurous sides have combined into a real love for science,” he says. Dr. Gormley currently channels his passion for science into his position as an assistant professor of biomedical engineering at Rutgers University in Piscataway, New Jersey.
Learning How the World Works
Both of Dr. Gormley’s parents worked in science and medicine—his mother as a medical doctor and his father as a physician-scientist—and they instilled in him a curiosity for how the world worked. When he was young, Dr. Gormley and his parents would tinker with cars or boats and fix broken household items together, all the while talking about the individual parts and how they functioned as a whole. “I always had that technical, hands-on side of me,” he says.
Continue reading “Using Robots and Artificial Intelligence to Search for New Medicines”

When she started college, Anne Carpenter, Ph.D., never guessed she’d one day create software for analyzing images of cells that would help identify potential medicines and that thousands of researchers would use. She wasn’t planning to become a computational biologist, or even to focus on science at all, but she’s now an institute scientist and the senior director of the Imaging Platform at the Broad Institute of Massachusetts Institute of Technology (MIT) and Harvard in Cambridge.
Starting Out in Science
Before beginning her undergraduate studies at Purdue University in West Lafayette, Indiana, Dr. Carpenter’s strongest interests were reading and writing. Then, her subjects expanded. “In college, I liked science as much as anything else, and I realized that was unusual, as a lot of other people really struggled with it. I decided to pursue science because I enjoyed it and the field had good job prospects,” she says. Dr. Carpenter majored in biology because she felt it had the “juiciest questions” as well as a direct impact on human health.
Continue reading “Automating Cellular Image Analysis to Find Potential Medicines”
The cloud. To many, it’s a mysterious black hole that somehow transports photos and files from their old or lost phone to their new one. To some researchers, though, it’s an invaluable resource that allows them access to data analytics tools they wouldn’t otherwise have.

Scientists have begun using cloud computing to store, process, and analyze their data through online bioinformatics tools. Biological data sets are often large and hard to interpret, requiring complex calculating instructions—or algorithms—to understand them. Fortunately, these algorithms can run on local computers or remotely through cloud computing.
One advantage of cloud-based programs over local computers is the ability to analyze data without taking up the user’s personal storage space. With cloud-based storage, researchers can store their large data files, including their labeled notes called annotations. Another benefit is that users have easy access to software packages within the cloud for data analysis. The cloud also encourages collaboration among scientists by making it easy to share large amounts of data.
Continue reading “Cloudy With a Chance of Scientific Discoveries” Dr. Brian Munsky. Credit: Colorado State University.
Dr. Brian Munsky. Credit: Colorado State University.
“I think having a career in science is really the best way to rechannel the inner child, to remain forever curious about the world,” says Brian Munsky, Ph.D., an associate professor of chemical and biological engineering at Colorado State University, Fort Collins. Check out the highlights of our interview with Dr. Munsky below to learn how his childhood practical jokes led to him running a research group that uses computational and experimental methods to study complex processes inside cells.
Continue reading “Career Conversations: Q&A With Biological Engineer Brian Munsky”
“You’re not going to be able to do biology without understanding programming in the future,” Melissa Wilson, Ph.D., an associate professor of genomics, evolution, and bioinformatics at Arizona State University, said in her 2019 NIGMS Early Career Investigator Lecture. “You don’t have to be an expert programmer. But without understanding programming, I can assert you won’t be able to do biology in the next 20 years.”
A growing number of researchers, like Dr. Wilson, are studying biology using computers and mathematical methods. Some of them started in traditional biology or other life science labs, while others studied computer science or math first. Here, we’re featuring two researchers who took different paths to computational biology.
Continue reading “Biology Beyond the Lab: Using Computers to Study Life”As computers have advanced over the past few decades, researchers have been able to work with larger and more complex datasets than ever before. The science of using computers to investigate biological data is called bioinformatics, and it’s helping scientists make important discoveries, such as finding versions of genes that affect a person’s risk for developing various types of cancer. Many scientists believe that almost all biologists will use bioinformatics to some degree in the future.
However, bioinformatics isn’t always included in college biology programs, and many of today’s researchers received their training before bioinformatics was widely taught. To address these gaps, the bioinformatics cores of the five Northeast IDeA Networks of Biomedical Research Excellence (INBREs)—located in Maine, Rhode Island, Delaware, Vermont, and New Hampshire—have worked together to offer basic bioinformatics training to students and researchers. The collaboration started in 2009 with a project where researchers sequenced the genome of a fish called the little skate (Leucoraja erinacea) and used the data to develop trainings.
Continue reading “Gone Fishing: Teaching Bioinformatics With Skate DNA”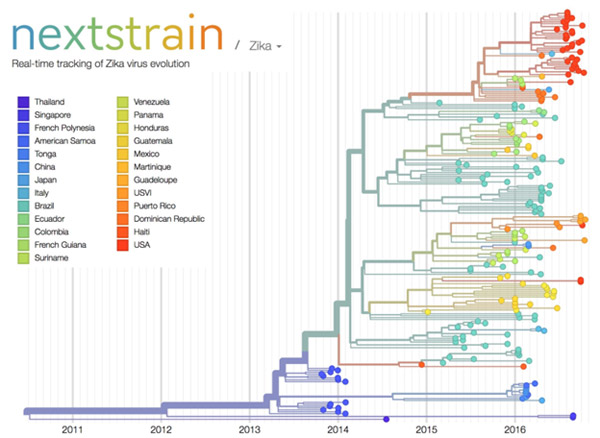
Over the past decade, scientists and clinicians have eagerly deposited their burgeoning biomedical data into publicly accessible databases. However, a lack of computational tools for sharing and synthesizing the data has prevented this wealth of information from being fully utilized.
In an attempt to unleash the power of open-access data, the National Institutes of Health, in collaboration with the Howard Hughes Medical Institute and Britain’s Wellcome Trust, launched the Open Science Prize. Last week, after a multi-stage public voting process, the inaugural award was announced. The winner of the grand prize—and $230,000—is a prototype computational tool called nextstrain ![]() that tracks the spread of emerging viruses such as Ebola and Zika. This tool could be especially valuable in revealing the transmission patterns and geographic spread of new outbreaks before vaccines are available, such as during the 2013-2016 Ebola epidemic and the current Zika epidemic.
that tracks the spread of emerging viruses such as Ebola and Zika. This tool could be especially valuable in revealing the transmission patterns and geographic spread of new outbreaks before vaccines are available, such as during the 2013-2016 Ebola epidemic and the current Zika epidemic.
An international team of scientists—led by NIGMS grantee Trevor Bedford of the Fred Hutchinson Cancer Research Center, Seattle, and Richard Neher ![]() of Biozentrum at the University of Basel, Switzerland—developed nextstrain as an open-access system capable of sharing and analyzing viral genomes. The system mines viral genome sequence data that researchers have made publicly available online. nextstrain then rapidly determines the evolutionary relationships among all the viruses in its database and displays the results of its analyses on an interactive public website.
of Biozentrum at the University of Basel, Switzerland—developed nextstrain as an open-access system capable of sharing and analyzing viral genomes. The system mines viral genome sequence data that researchers have made publicly available online. nextstrain then rapidly determines the evolutionary relationships among all the viruses in its database and displays the results of its analyses on an interactive public website.
The image here shows nextstrain’s analysis of the genomes from Zika virus obtained in 25 countries over the past few years. Plotting the relatedness of these viral strains on a timeline provides investigators a sense of how the virus has spread and evolved, and which strains are genetically similar. Researchers can upload genome sequences of newly discovered viral strains—in this case Zika—and find out in short order how their new strain relates to previously discovered strains, which could potentially impact treatment decisions.
Nearly 100 interdisciplinary teams comprising 450 innovators from 45 nations competed for the Open Science Prize. More than 3,500 people from six continents voted online for the winner. Other finalists for the prize focused on brain maps, gene discovery, air-quality monitoring, neuroimaging and drug discovery.
nextstrain was funded in part by NIH under grant U54GM111274.
Do you like to find new uses for old things? Like weaving old shirts into a rug, repurposing bottles into candle holders or turning packing crates into tables? Katie Gostic, a University of California, Los Angeles (UCLA) graduate student, likes finding new uses for old data. She channeled this interest when she analyzed existing data to study whether childhood exposure to flu affects a person’s future immunity to the disease.

As an undergraduate student at Princeton University, Gostic was originally pursuing a degree in engineering. Her focus shifted to biology after taking an infectious disease modeling class. Gostic’s background in math and programming allows her to take large, complex pre-existing data sets and reanalyze them using new tools and methods. The result: Information that wasn’t accessible when the data were first collected.
Now a graduate researcher in the ecology and evolutionary biology lab of James Lloyd-Smith ![]() , Gostic studies infectious diseases. The lab builds mathematical models to investigate zoonotic diseases—diseases that animals can transmit to humans but that humans don’t frequently spread between each other. Examples include diseases caused by Leptospira, a type of bacteria that infects household pets and many other animals, and monkeypox, a virus whose transmission to humans is increasing since the eradication of smallpox. The lab also studies bird flus, a category of flu viruses that infect birds and other animals and only occasionally jump to people. A very small number of cases of human-to-human transmission of bird flus have been recorded. However, if a bird flu virus mutated in a way that allowed it to spread among humans, it could cause a pandemic. Continue reading “Student Researcher Finds New Clues About Flu with Old Data”
, Gostic studies infectious diseases. The lab builds mathematical models to investigate zoonotic diseases—diseases that animals can transmit to humans but that humans don’t frequently spread between each other. Examples include diseases caused by Leptospira, a type of bacteria that infects household pets and many other animals, and monkeypox, a virus whose transmission to humans is increasing since the eradication of smallpox. The lab also studies bird flus, a category of flu viruses that infect birds and other animals and only occasionally jump to people. A very small number of cases of human-to-human transmission of bird flus have been recorded. However, if a bird flu virus mutated in a way that allowed it to spread among humans, it could cause a pandemic. Continue reading “Student Researcher Finds New Clues About Flu with Old Data”