DNA, with its double-helix shape, is the stuff of genes. But genes themselves are only “recipes” for protein molecules, which are molecules that do the real heavy lifting (or do much of the work) inside cells.
 Artist interpretation of RNAP grasping and unwinding a DNA double helix. Credit: Wei Lin and Richard H. Ebright.
Artist interpretation of RNAP grasping and unwinding a DNA double helix. Credit: Wei Lin and Richard H. Ebright.Here’s how it works. A molecular machine called RNA polymerase (RNAP) travels along DNA to find a place where a gene begins. RNAP uses a crab-claw-like structure to grasp and unwind the DNA double helix at that spot. RNAP then copies (“transcribes”) the gene into messenger RNA (mRNA), a molecule similar to DNA.
The mRNA molecule travels to one of the cell’s many protein-making factories (ribosomes), which use the mRNA message as instructions for making a specific protein.
Because making proteins is essential for life, so is RNAP. All living organisms—from humans down to bacteria—have RNAP.
Due to its vital role, RNAP also makes an excellent target for medicines. Drugs that interfere with the form of RNAP present in bacteria (but not the form of RNAP present in humans) can effectively kill bacteria while leaving the human RNAP intact, making them great candidates for treating infections.
Richard H. Ebright, a scientist at Rutgers, The State University of New Jersey, is studying RNAP to learn how it copies information in DNA. His goal is to find new ways to kill bacteria, particularly bacteria that are resistant to current antibiotics. He asks questions such as:
- How does RNAP find the right place to start copying the recipe for a protein?
- How does RNAP grasp and unwind the DNA double helix to start copying the recipe?
- How does RNAP move along DNA as it copies the recipe?
- How do drugs that inhibit bacterial RNAP work?
- Can we find new drugs that inhibit bacterial RNAP?
The RNAP-DNA connection is extremely fast and nearly invisible, even using sensitive equipment, so scientists are still seeking answers to many of these questions. But researchers have made a lot of progress. In particular, they now have a much better idea of how various antibiotics stop RNAP from working.
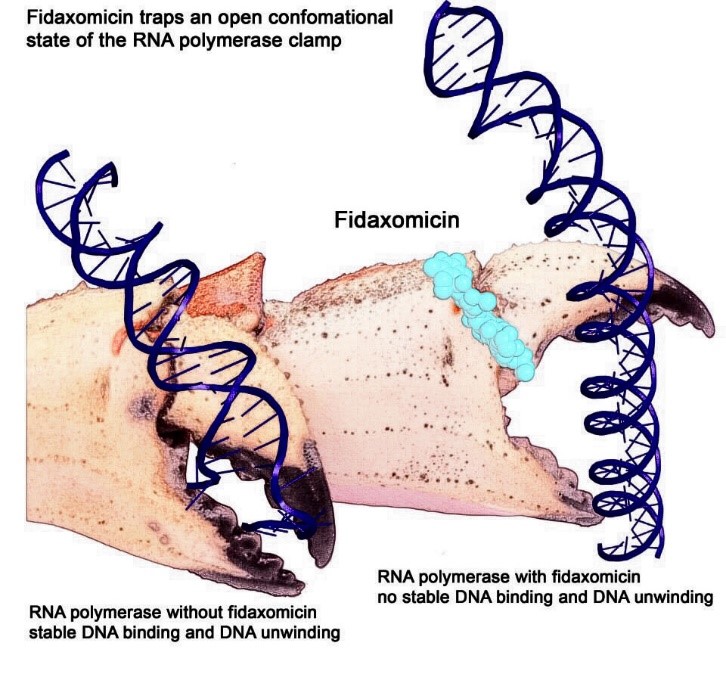
In a 2018 study, for example, Ebright and his team first described how fidaxomicin, a new antibiotic, was able to fight formidable bacteria such as Clostridium difficile. The key was its ability to stop RNAP. Fidaxomicin works by binding to RNAP at the base of one of the two pincers of the RNAP claw. This prevents the claw from grasping and unwinding DNA.
Credit: Wei Lin and Richard H. Ebright.A year earlier, in 2017, Ebright and his team examined another antibiotic, called pseudouridimycin (PUM), which is produced by a microbe isolated from a soil sample collected in Italy. They showed that PUM kills bacteria by mimicking a chemical building block called uridine triphosphate. RNAP uses that building block to make RNA, so it’s a key component of the instructions for the final protein. PUM prevents RNAP from making RNA by taking the place of uridine triphosphate and preventing it from binding to RNAP.
In another 2017 study, Ebright and his team looked at the antibiotic rifampin. Rifampin has always been essential for the treatment of tuberculosis but, over time, tuberculosis bacteria have become increasingly resistant to this antibiotic. The scientists traced this resistance to RNAP. They found that tuberculosis RNAP has learned how to make changes to prevent rifampin from binding to it, rendering the drug less effective. However, at the same time, the researchers found potentially new types of drugs, called AAPs (Nα-aroylN-aryl-phenylalaninamides), that kill tuberculosis bacteria by binding to a different, completely separate, binding site on RNAP. Because AAPs target this new site, they are proving effective, even against rifampin-resistant tuberculosis bacteria.
Even with promising stories such as these, the battle against disease-causing bacteria is far from over, and RNAP research will be a big part of this next chapter. “Bacteria will always acquire resistance,” Ebright says. “But for large, complex protein machines such as RNA polymerase that carry out complex tasks, there are many ways to interfere with their function and, thus, many opportunities to find new drugs.”
Ebright’s research is supported in part by NIGMS under grant number R37GM041376.
Very informative and clear tutorial on how antibiotics target the action of RNAP